




医疗器械博览会解读FDA医疗器械设计和开发控制指南
2023-07-28
医疗器械承担着巨大的责任和风险——如果它们不能正常工作,可能会对人的健康和生命造成非常严重的后果。因此,医疗器械在美国受到食品和药物管理局(FDA)的严格监管。
我们看下该指南的适用范围:FDA对医疗设备制造商的设计控制适用于所有第二类和第三类设备以及图中所示的第一类器械:
(1)有计算机软件实现自动化的器械;
(2)支气管引流管、外科医生手套、防护性限位器、手控放射性核素置位系统、放射性核素远程治疗源器械。

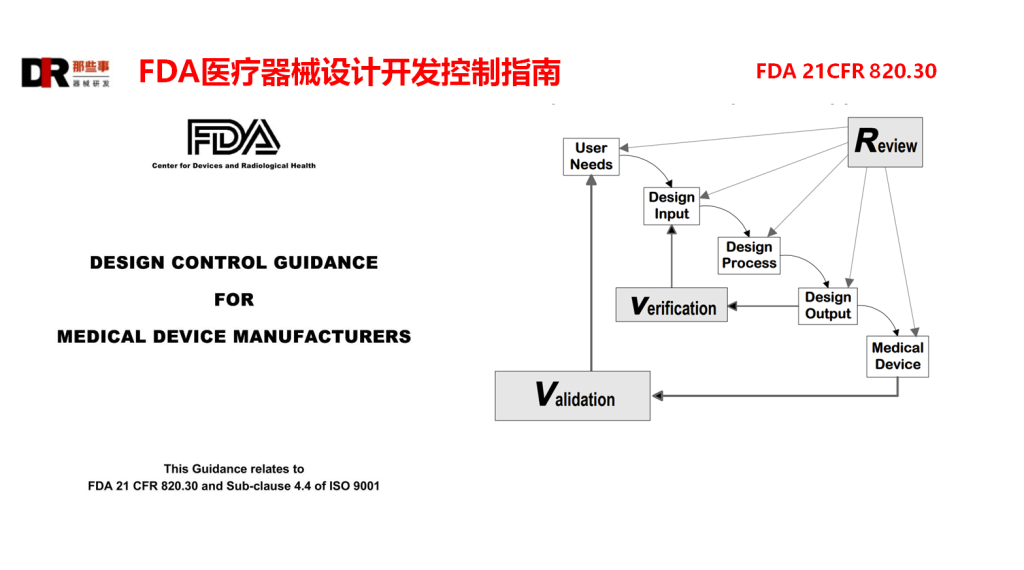
接下来我们看下设计开发的阶段,其实FDA的阶段与之前介绍ISO13485或GB/T 42061-2022中的阶段大同小异。在此主要介绍一下区别的地方。对于设计和开发策划阶段基本是一致的,对于设计输入阶段明确了对于不完整、含糊或有相互矛盾需求的处理机制的要求。对于设计评审增加了无直接责任人和专家的要求。

对于设计验证、设计确认、设计转换和设计更改的要求基本是一致的。不同的是ISO13485或GB/T 42061-2022仅要求了设计开发文档,但并没有对设计开发文档给出明确的要求和类型划分。FDA的指南对此给出了明确的区分和划分,以及要求。

具体而言,设计开发文档包括设计历史文档(Design history file ) 和器械主记录(Device master record),也就是我们经常容易混淆的DHF和DMR。我们先了解下DHF,原文中Design history file (DHF ) means a compilation of records which describes the design history of a finished device.简单来说,DHF是回答器械如何设计出来的过程文档。
设计历史文档(DHF ) :指的是描述某医疗器械成品设计过程的有关记录。
包含内容:DHF包括设计开发活动的所有记录。
从最初的策划、客户需求、设计输入,设计输出、设计验证、设计确认、所有的设计评审、设计转换、以及设计变更相关的所有记录。均属于DHF的一部分。
要求:各制造商应为每个类型的器械建立并保持设计历史文件。设计历史文件应包含或引用必要的记录,以证实设计符合经批准的设计计划和法规 的要求。
参与者:设计开发过程涉及的所有人,包括但不局限于:市场部、研发部、质量部、生产部、 临床部、注册部等。
曾在医疗器械博览会中亮相的研发和设计服务企业,包括上海威固、上海药明康德、巨翊科技、上海瀚赛、诺达思(北京)信息技术、北京卓杰亿品、洛可可创新设计集团等多家企业,他们在现场带来展品包括OSSD-BGA SATA nano 、医疗器械测试业务、临床前大动物实验、行为观察记录分析系统等。点击快速加入医疗器械博览会。

器械主记录(DMR ) :DMR means a compilation of records containing the procedures and specifications for a finished device.DMR是指包括医疗器械成品的程序和规范的完整记录。简单来说,DMR是回答器械如何制造出来所需要的文档。
包含内容:DMR包含了制造和测试医疗器械所需的一切程序和规范。
每类器械的 DMR 应包括下列信息或指明所在位置 :
(a)器械规范包括相应的图纸、组成、配方、组件规范和软件规范
(b)生产加工规范包括相应的设备规范、生产方法、生产程序、生产环境规范
(c)品质保证程序和规范,包括接收标准和使用的质量保证设备
(d)包装和标记规范,包括使用和处理方法
(e)安装、维护和服务的程序及方法。
要求:各制造商应保留器械主记录(DMRs),并保证每个器械主记录都按照“文件控制” 的要求进行准备和批准。
参与者:研发部、生产部、质量部等相关部门

由于这两类文档常常被大家混淆,我们看下这两者之间的区别是什么。首先我们从DHF和DMR的英文全称就可以发现其对象是不同的,DHF中的D是Design,是为了证明这一类产品的设计是符合设计计划及法规要求的。大部分设计开发文档DHF于产品上市前完成,并根据设计开发文档准备产品的注册资料,以证明医疗器械符合安全性和有效性的要求,取得上市许可。因此绝大部分注册资料都是来源于DHF。上市许可后对产品的任何设计变更,其相关研究及记录均属于DHF的一部分。有些设计变更需要取得主管部门的许可。如前所述,DHF是回答器械是如何设计出来的。
DMR中的D是Device。在证明产品设计没有问题后,DMR的作用是为了保证能够持续稳定地制造出符合接受标准的器械。有一些DMR文件包含在DHF文件中,如:
-
器械规范(技术要求)、包装和标签属于设计输出的一部分;
2. 生产工艺规范属于设计转换的一部分;
3. 质量保证程序和规范也是在设计开发过程中创建的,因为其中包括作为设计输出一部分定义的接受标准。通俗讲包括原材料/半成品/成品检验规程;生产环境的要求及控制规范等。

我们看下FDA设计和开发指南的精髓。其介绍了传统瀑布模型(waterfallmodel)和同步工程模型(concurrent engineeringmodel)的区别,同步工程模型更适用于复杂的器械开发,详细的可查看原文的内容。同时该指南模糊研发和生产的界限,研发时就要考虑量产的要求(DFM),也就是说生产工艺的研究属于设计开发范畴。重视设计开发的程序和文档,包括风险管理、器械可靠性、器械耐用性、器械可维修性、器械可服务性 人因工程、软件工程、使用标准、配置管理、法规要求的符合性、器械评估(可能包括第三方产品认证或批准)、临床评估、文件控制、使用顾问、使用分包商、使用公司历史数据等。

此外,将风险管理用于设计开发 也是该指南的精髓之一。风险管理被规定为一个框架,这个框架内,经验,洞察和判断被成功地应用于管理风险。风险管理在设计输入要求的开发开始启动。随着设计的进展,新的风险可能成为证据。为了系统的识别并在必要时降低这些风险,风险管理过程是并入到设计过程中的。通过这种方式,能够在设计过程中较早的时期识别出并管理不可接受的风险,这个时期进行变更会较容易且花费也会相对较低。
举例:X射线系统的曝光控制系统,控制功能是配置在软件上的。开发过程后期,这个系统的风险分析没有设计一些可能导致患者过度曝光的失败模型。这是因为这个问题直到设计将要结束的 时候才被识别出来,需要增加一个昂贵的、单独的备用计时器来管理曝光时间。
医疗器械博览会意识到,由于风险管理是贯穿整个产品生命周期的,要看下风险管理的3重境界:已知风险、内部风险、未知风险。在此特别提一下内部风险,其是影响产品开发周期最为严重的一个因素,往往很多时候我们无法识别或无法取舍,会对注册周期产生较大影响。

文章来源:





