




印度医疗器械监管重点
2018-06-06
印度医疗器械市场位居全球前20,同时也是亚洲第四大医疗器械市场。其市场规模大约在55亿美元左右,预计复合年增长率为15%。
印度的医疗器械市场在过去的二十年中经历了持续不断的变革。在1991年的“新经济政策”之前,印度的医疗器械在国内制造业中占据着主导地位。之后,它变成了一个以进口产品驱动的市场。在2006年之前,印度的医疗设备部门都并没有受到监管;直到2006年,印度中央药品标准控制组织(CDSCO)通知了15类医疗设备需要进行注册,这样的时代才算宣告结束。
为了配合落实“印度制造”计划,CDSCO发布了新的《医疗器械法规2017》(Medical Device Rules,2017),该法规于2018年1月1日正式开始实施。在实施《医疗器械法规2017》之前,根据《药品和化妆品法案1940》(Drug and Cosmetic Act, 1940),在印度医疗器械都是按照药品标准受到监管的。因此,有必要将药品和医疗器械区分开来。其次,也迫切需要为当地制造商在印度发展行业提供更有利的环境。最后,工商部在2017年发布了公共采购方案,并将药品部门确定为公告机构。
制定新法规是为了促进国内制造业的发展并且规范该地区的进口和制造业。目前,跨国公司占据了印度医疗器械市场75%的销售额。新法规遵循了GHTF(全球协调工作组)的指南并且与其中基于风险的分类方法一致。此外,公告机构的检查也被引入到了新的医疗器械法规中来。本文强调了一些要点以便于更好地理解《医疗器械法规2017》。
分类
与全球法规一致,新法规引入了基于风险的分类系统。CDSCO将医疗设备进行了分类,并在其网站上不时发布分类的设备列表。进口商和制造商必须按照分类表对他们的设备进行分类。如果一个设备在GHFT国家的分类等级更高,那么将对其考虑更高级别的分类。
《医疗器械法规2017》中的设备分类系统
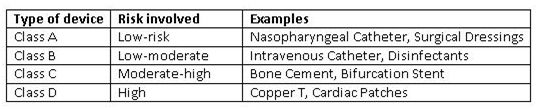
质量管理体系(QMS)评估
根据新法规,一种经过公告机构“第三方合格评定和认证”新程序被引入。公告机构可以在生产场所对A类和B类设备执行QMS评估。根据要求,公告机构也可以支持CDSCO为C类和D类医疗设备的生产现场进行QMS评估。经认证的机构名单将会在其网站上公示。对于国外制造商,CDSCO也可能会要求由CDSCO内部检查员或者任何公告机构对其海外生产基地进行检查。
注册
新法规将强制要求所有设备获得制造和进口许可证。所有制造和进口许可证的申请都是通过在线门户网站SUGAM来处理的,SUGAM是一个隶属于印度卫生和家庭福利部(Ministry of Health and Family Welfare)的在线授权系统。
印度国家授权许可机构(SLA)将对A类和B类设备的制造许可进行监管,而C类和D类许可申请将提交给中央授权许可机构(FSSAI)。质量评估报告(QAR)必须与B类、C类和D类设备的制造许可证申请一同提交。而A类医疗设备的QAR只需要在制造许可证颁发之日起120天内提交即可。
Requirements for approval of product
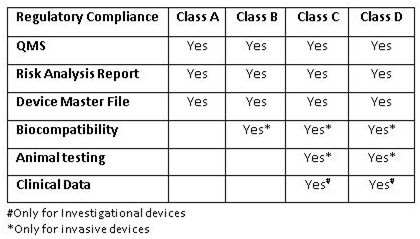
要获得进口许可证,需要先获得制造或分销许可证。国外制造商应指定一名获得授权的印度代理商来持有许可证,才能进行上市后监察(post marketing surveillance,PMS)活动和医疗设备的分销。所有类别的医疗器械进口许可证申请都必须提交中央授权许可机构。
与制造许可证不同的是,进口许可证的申请不需要进行QAR,但如果需要,中央授权许可机构可能会检查国外的场地。为了保证新的指导方针更加严格,进口商现在必须向中央授权许可机构提交完整的技术文件和进口许可证申请,并且每个印度代理商将负责在国内的PMS活动。根据2017年的新法规,相同产品的多个进口许可证可以由不同的印度代理商代理。
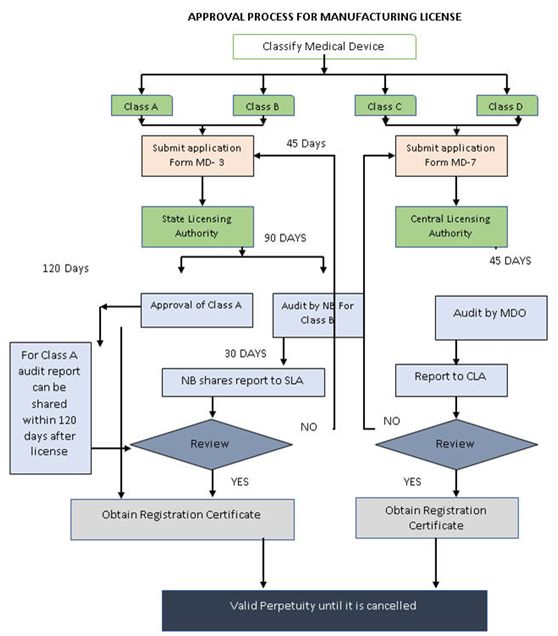
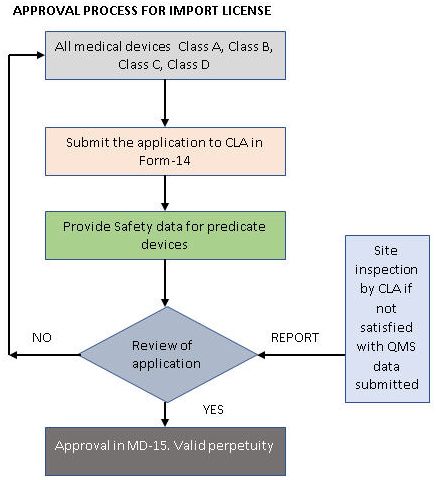
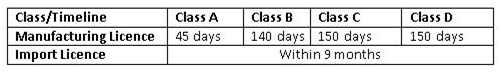
所有获得授予的许可都是永久性的,除非它们被取消。为了保留许可证,持有人每五年需要支付一次执照留置费。印度政府特别注意到了时间表是不可避免的,因此合理化设置了授予许可证的时间。
获得医疗器械制造/进口许可证的时间
<
临床研究
《医疗器械法规2017》改变了医疗器械临床试验方案,将其从一个与药物相同的四期试验改成了两期试验。这两期将分为实验性临床研究(探索性研究)和关键临床研究(验证性研究)。除此之外,PMS还必须在获得市场批准后才能进行。然而,对于一种在印度还没有已获批准类似器械的医疗器械申请进口许可证,如果已经获得澳大利亚、加拿大、日本、美国或欧盟成员国机构当局颁发的自由销售证书(FSC),就不需要进行临床研究。
此外,在新法规中引入了用于医疗器械(除科研型医疗器械)已获批准类似器械的“实质等同性原则(Substantial Equivalence)”。对于体外诊断产品,“临床表现评估”将成为监管要求的一部分。此外,临床试验所产生的数据如果不是用于申请制造或进口许可证的话,将不需要事先获得临床试验批准。
标签
根据新法规中的规格说明,标签要求被强制性要求遵守。除此之外,印度政府还强制执行了《法定计量(包装商品)法规2011》。此外,每个设备的医疗器械唯一标识(UDI)号码必须在标签上注明,有效期至2022年1月。
召回
药品和化妆品法案不能强制制造商或进口商从市场上撤回产品。而根据新法规,制造商或进口商必须召回任何危险或有害的产品,并要求他们提供召回的理由。
公共采购订单(PPO)
为了改善国内制造商生态系统,印度政府(GoI)为公共采购订单(PPO)制定了准则草案。这些规定覆盖了价值50 lakh或以下的公共采购医疗设备的标书。当地成分(local content)成本的确定应以人力和材料(主要成分)的原产国为基础。政府提出了一个计算当地成分的公式:
D =(A / C)* 100
(C = A + B)
D=当地成分的百分比
C =总成本
B=进口部件成本
A=国产部件成本
Minimum local content in domestic medical devices fixed by GoI
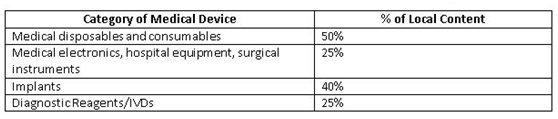
结论
《医疗器械法规2017》有着许多能够鼓励印度医疗器械行业发展的特点。通过引入单个在线门户,注册流程已经被简化。公告机构的审计将进一步提高设备制造的质量。临床试验要求的改变将鼓励新医疗设备的创新。这些规定将鼓励国产制造,并加强对进口许可证文件的审查。
原作者:Harish Reddy Arkala, Freyr
原文链接:
https://www.meddeviceonline.com/doc/medical-devices-regulatory-priorities-in-india-0001




