




政策法规变动对医疗器械产业的影响
2018-04-26
当前,以新版《条例》为核心,以部门规章为主体,以规范性文件为补充,我国已经构建了覆盖医疗器械监管全过程的医疗器械法规体系。这场大规模的法规巨变和制度更新带给医疗器械行业什么影响?相关企业应该何去何从?本文试就这一问题进行探讨。
1、医疗器械政策法规正在经历巨变
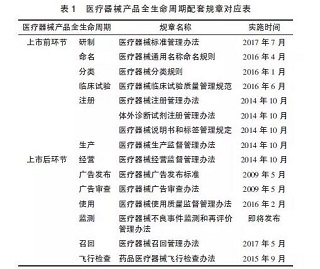
我国医疗器械法规体系的构建始于2000年《条例》的实施,经过2014年、2017年的2次修订后,法规体系的框架已经基本确立。根据产品全生命周期管理的需要,管理部门对相关配套规章做了较大的修改,基本上把原有医疗器械法规体系的内容回炉重造了一遍(医疗器械全生命周期配套规章见表1)。同时,还发布了《创新医疗器械特别审批程序》《医疗器械优先审批程序》《医疗器械生产质量管理规范》《医疗器械经营质量管理规范》等多部规范性文件。另外,为了推动这些管理规范的实施,CFDA以各种通知、公告、通告等形式发布的规范性文件就近百项。这些立法工作构建了行政法规、部门规章、规范性文件为主体框架的三级监管体系。这一切都在说明医疗器械政策法规正在经历着一场巨变。
2、政策法规变动对医疗器械产业发展的影响分析
根据Evaluate Med Tech的统计,2015年全球医疗器械销售规模为3903亿美元,2011-2015年全球医疗器械销售规模稳步增长,复合年均增长率(compound annual growth rate,CAGR)为1.90%。基于120家医疗器械行业内领先公司的数据测算,2015-2020年世界医疗器械产业复合年均增长率将达到5%,到2020年市场规模达到5140亿美元。根据此增速测算,2016年全球医疗器械销售规模为4063亿美元。而中国医疗器械产业的发展更令世界瞩目。进入21世纪以来,国内产业整体步入高速增长阶段,销售总规模从2001年的179亿元到2014年约2556亿元,增长了14.28倍,国内医疗器械市场规模增速大大高于全球,成为仅次于美国的全球第二大医疗器械市场。国内医疗器械2015年市场总规模约为3080亿元,2015-2020年医疗器械市场将保持20%的增速。根据此增速测算,2016年国内医疗器械销售规模为3696亿元,国内医疗器械市场规模大大高于全球增速,预计2017年将达到4942.3亿元,复合年均增长率预计为15.4%。
2.1对产业企业规模的影响
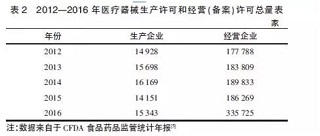
医疗器械是一个严格监管的产业,对生产经营主体均有资质要求,法规的修订会对产业规模产生明显影响,主要表现在获得资质的主体数量变化上。2014年《条例》首次修订后,要求生产第二类、第三类医疗器械的企业应该取得《医疗器械生产许可证》,要求经营第二类医疗器械的企业应该取得经营备案凭证,经营第三类医疗器械的企业应该取得《医疗器械经营许可证》。从表2可知,2012年到2014年持证经营的企业数量,由于法规没有大的修改,不管是医疗器械生产企业还是经营企业,均呈缓慢增长状态。
2015年,因为新版医疗器械药品生产质量管理规范(goodmanufacturing practices,GMP)和首版医疗器械药品经营质量管理规范(good supply practice,GSP)在该年的推行,医疗器械生产企业和经营企业数量均有减少,一些不符合医疗器械GMP要求的生产企业以及不符合医疗器械GSP要求的经营企业被淘汰出局,表明新政对行业产生了立竿见影的影响。
2016年,生产企业以及经营企业总量均有增加,说明新的规则被熟悉之后,更多的社会资源继续流入医疗器械这一朝阳产业。
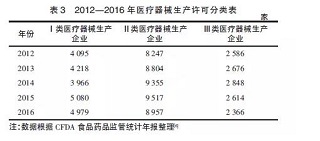
从表3中可以更深入地了解到新的政策法规实施对不同类别医疗器械生产企业的影响情况。从2012到2015年,从事第一类、第二类医疗器械生产活动的企业总体上处于增长状态。而对生产第三类医疗器械的厂家而言,2012-2014年每年都有小幅增长,但在2015、2016年均有减少。这与2015年3月国家开始要求第三类医疗器械生产企业全面执行医疗器械GMP 有直接关系,生产质量体系不合规且整改不合格的企业直接被淘汰。
另外,我国从2015年9月开始实施《药品医疗器械飞行检查办法》。管理部门利用飞行检查手段,对医疗器械生产企业展开了密集的不预先告知的行政检查。2016年,所有类别的生产企业数量均有所下降,这跟当年严厉的飞行检查淘汰劣质企业不无关系。
2.2 对产品备案和注册的影响
医疗器械产品上市状况是行业发展的“晴雨表”,新实施的政策法规也直接影响着产品的备案和注册数量。如表4所示,2014年《条例》将第一类医疗器械产品注册管理调整为备案管理后,备案产品有了大幅上升,增长率达到63.5%。第一类医疗器械备案率的大幅增长,主要是由于第一类医疗器械的管理方式发生了根本变化,从注册管理改为备案管理,刺激了第一类医疗器械的增长。
同时,第二类医疗器械的首次注册数,从2015年的7029件下降到了2016年的6093件,降幅达到13.31%;进口医疗器械的首次注册数,从2015年的1559件降低到了2016年的1037件,降幅达到33.48%。2项数据的下降表明2016年开展的医疗器械临床试验数据真实性核查行动对企业的首次注册申报产生了实质的影响。另外,2015年开始实施的医疗器械注册收费制度也使企业的注册申请更为谨慎,影响了医疗器械生产企业的注册申请数量。在这些政策影响下,企业的备案与注册行为更为规范化,提升了产品备案与注册申报的质量。
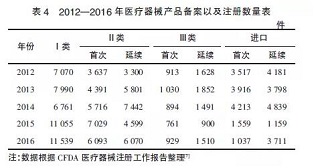
值得注意的是,2016年体外诊断试剂异军突起,在注册产品中占的比重很大。从CFDA公布的注册数据看,境内第三类医疗器械中,体外诊断试剂注册数量占全部注册数量的43%;进口医疗器械中,体外诊断试剂注册数量占全部注册数量的33%。这些都表明,在医疗器械行业版图中,体外诊断试剂所占的份额将越来越大。
我国于2014年3月开始实施《创新医疗器械特别审批程序》,允许符合条件的创新医疗器械进行特别审批。这一政策的出发点是缩短创新医疗器械的注册审评时间,以加快产品的上市步伐。但是,创新医疗器械产品属于高科技和学科交叉产品,风险性高,技术审评难度大,导致审评周期时间长,效率偏低。由于创新医疗器械的门槛要求高,再加上审评慢,致使该政策不能从根本上促进医疗器械行业的创新发展。
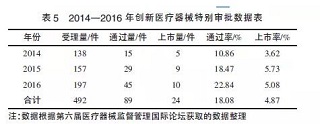
从表5可以看出,实施创新医疗器械特别审批程序后,创新医疗器械的通过量和上市量在2014-2016年均稳步增长,但由于申报量越来越大,上市率稍有起伏。2015年,CFDA批准注册脱细胞角膜基质等9个产品上市(其中有源医疗器械5项,体外诊断试剂3项,无源医疗器械1 项)。2016年,批准注册三维心脏电生理标测系统等10个产品上市(其中有源医疗器械6项,无源医疗器械3项,体外诊断试剂1项)。2016年,不管是创新医疗器械的注册申请量,还是经过审评后的通过量以及最终获批的上市量都有所上升,说明创新医疗器械特别审批这一新政继续推动着产业技术和产品的更新。
从以上分析可知,医疗器械产业对政策与法规的变动相当敏感。相关政策和法规稍有变动,将引起行业产业的直接反应。由于医疗器械与人们的生命安全和身体健康密切相关,国家必须对它实施最为严厉的监管,这对产业发展的影响无疑是深远的。
2.3 对产业发展环境的影响
经过大规模的政策法规变动之后,医疗器械产业发展环境也会发生很大变化。这种影响变化主要体现在制度环境变严和产业环境趋活2个方面:
(1)经过政策法规的调整,医疗器械管理制度的“笼子”已经编织严密,从严监管成为产业管理主轴,制度环境变严。2017年9月1日,《最高人民法院、最高人民检察院关于办理药品、医疗器械注册申请材料造假刑事案件适用法律若干问题的解释》开始正式实施。该项司法解释将药品和医疗器械临床试验数据造假纳入刑法处理,在刑罚层面进一步强化了行业的从严监管。该项司法解释规定,药物非临床研究机构、药物临床试验机构、合同研究组织、医疗器械临床试验机构、药品医疗器械注册申请单位、药品医疗器械监管部门及其工作人员故意提供虚假的医疗器械临床试验报告及相关材料,情节严重的,以提供虚假证明文件罪追究刑事责任。除此之外,管理部门频繁的飞行检查、“双随机”检查、抽验检查以及临床数据真实性核查等行政检查活动,也使行业的制度环境变严了。
2017年5月4日,国务院发布了关于修订《医疗器械监督管理条例》(以下简称《条例》)的决定,这已经是这部医疗器械行业“母法”在短时间内的第二次修改,引起了医疗器械行业企业的极大关注。2014年,《条例》第一次修订后,管理部门大幅更新了配套的部门规章,从而使医疗器械全过程各个环节均有法可依。
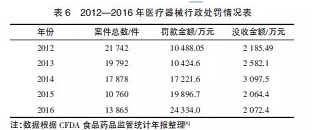
从表6可知,2012-2016年,行政处罚案件总数起伏不定。2015年查处的案件总数发生剧降,2016年有所回升。这表明《条例》修订后,由于不熟悉新的罚则,管理部门查处违法行为的行动力有所减弱,但后期增强了。从2013年起,罚款金额作为一个主要的处罚指标一直稳步上升,表明对医疗器械的监管力度和处罚力度仍在持续加强。由于法规加大了处罚力度,因企业生产经营方面的合规问题被罚高达近千万元的案例并不罕见。这是广大医疗器械生产企业所不能懈怠的。
(2)系列关于鼓励行业创新发展的利好政策的出台,给行业带来新的发展机遇,产业发展环境趋向活跃。除了《创新医疗器械特别审批程序》这一政策将持续推动产业技术与产品的创新之外,2017年1月开始实施的《医疗器械优先审批程序》,将加快那些治疗罕见病、恶性肿瘤有明显临床优势或列入国家科技重大专项、国家重点研发计划的器械上市步伐。
2017年5月11-12日,CFDA连续发布了2017年第52、53、54、55号等4个关于鼓励药品医疗器械创新政策征求意见稿的公告,体现了药品医疗器械管理的改革趋势。4份征求意见稿中,与医疗器械密切相关的政策动态主要有以下几点:一是要建立医疗器械审评团队负责创新类医疗器械审评并实施项目管理人制度,这将直接提高当前创新医疗器械的审评效率,并加强其审评的公正性;二是将医疗器械临床试验机构资格认定改为备案管理,鼓励社会资本投资成立临床试验机构,完善伦理委员会机制并接受境外临床试验数据用于医疗器械注册审评,这既给改善当前医疗器械临床试验机构不足的窘境提供了方法途径,又使一些拥有境外临床试验资源的企业能够抢占注册审批先机;三是将药品上市许可持有人制度实施的经验推广到医疗器械注册环节,完善医疗器械不良事件报告和再评价制度,建立职业化的医疗器械检查员队伍等,未来医疗器械的生产与注册将进一步解绑,以放宽医疗器械注册申请人的诸多限制,从而刺激行业创新发展。
2017年10月8日,中共中央办公厅、国务院办公厅印发了《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》,为解决以上问题做出了顶层设计,一些长期困扰医疗器械行业创新发展的瓶颈将会打破,这将对医疗器械产业的创新发展产生深远而又重大的影响。
3、政策法规变动下医疗器械产业发展的建议
从上述分析可知,医疗器械政策法规目前正处于变动期,并呈现出明显的严厉趋势。在日益趋严的产业环境中,政策法规的灵活性给行业带来了发展机遇。未来,企业既要下大力气做好质量合规,以便满足严格的监管要求;又要善于利用政策红利,捕捉难得的发展机会。
(1)把握医疗器械政策法规发展方向顺势而为。
《关于深化审评审批制度改革鼓励药品医疗器械创新的意见》反映了产业管理的最新发展动向,企业应该从中准确把握医疗器械政策法规发展的趋势,及早布局筹划新的发展规划。尤其是关于医疗器械注册审评的改革动向,企业应该保持密切关注。
(2)做好医疗器械上市前质量合规管理。
在从严监管的行业环境下,企业必须扎实履行好法规规定的法律义务,尤其是要做好医疗器械产品上市前的质量合规管理。企业应该在研发、检验、临床试验、注册以及生产等上市前环节里,不折不扣履行法律法规规定的义务,确保质量管理体系有效运行。2017年新修订的《条例》已经将医疗器械临床试验机构的资质认定改为备案管理,这已经在立法上先于药品确定了临床试验机构的备案管理制度。未来将有更多符合条件的机构能够提供专业的临床试验服务,这对于医疗器械的质量合规大有裨益。但如前所述,对于在医疗器械注册申请过程中材料弄虚作假的情形,情节严重的,可能追究刑事责任。严厉的责任规定,应该成为企业不可触碰的红线。
(3)履行医疗器械上市后企业主体责任。
未来,基于产品全生命周期管理的需要,管理的重心将继续从产品上市前审批向上市后监测转移,从而也要求企业在产业发展全程中承担一以贯之的法律义务。医疗器械生产企业以及使用单位应当按照上市后产品的不良事件监测、再评价以及召回的相关规定,积极申报不良事件和召回缺陷产品。企业应加强自律,严格把关产品质量,防止不良事件的发生对产业造成打击。医疗机构应对在用医疗器械做好集中管理,使其把有限的人力、财力、精力都集中到医疗技术上,提升内涵、持续创新、提高诊疗水平和质量,使医疗技术更富含金量。
(4)实施行业产业差异化发展策略。
当前,医疗模式正在从过去的诊断—治疗模式向着预防—诊断—治疗—康复—保健模式转变。在此背景下,医疗需求将呈现新的趋势,家用医疗仪器、家用辅助类机器人设备、社区康复用医疗设备、面向电子健康档案的技术与产品将会成为新的热点产品领域。在新的法规和政策环境中,3D 打印医疗器械、移动医疗设备、基因测序、家用医疗器械等细分行业应该抓住机遇,以差异化发展策略取得竞争优势。
4、结语
2014年以来,我国医疗器械政策法规发生的巨变改变了产业发展的环境,医疗器械产业经历了制度和行业的双重分水岭。在医疗器械政策法规体系得到长足发展的背景下,产业从严监管的主题将在未来长期坚持不变。在这一政策法规巨变背景下,医疗器械产业也面临着难得的发展机遇。如果企业能够顺应法规的变化,严格在法律的框架下开展生产经营活动并履行好自身的主体责任,注重技术与产品的创新并实施差异化竞争的策略,必将能推动产业发展迈向一个新台阶
来源:医谷




