




1.项目概述
作为2014~2015 战略重点,FDA 分两阶段实施了该项目,其计划和完成情况如下。
1.1 第一阶段
计划:到2014 年12 月31 日为止,完成50% 已上市PMA 器械的回顾性审核,以确定是否需要将一些上市前数据要求更改为上市后相关设定或进行重新分类,并将相关决定向公众发布[1]。
结果:在2014 年,CDRH 完成了69% 已上市PMA 器械的回顾性审核。
1.2 第二阶段
计划:到2015 年12 月31 日为止,完成全部已上市PMA 器械的审核,已确定是否需要将一些上市前数据要求更改为上市后相关设定或进行重新分类,并将相关决定向公众发布[2]。
结果: 在2015 年,CDRH 完成了剩余部分已上市PMA 器械的回顾性审核。
2.调整方案
FDA 官方网站分别于2015 年4 月和2016 年1 月对审核结果进行了在线发布。在其发布的审核结论中,分为四种处理思路:
(1)拟降为Ⅱ管理(见表1):2014 年确定拟降为Ⅱ类的共21 项,到2017 年1 月为止,仅唾液系统刺激器(LTF)于2015 被重新分为Ⅱ类,其他尚未发布重新分类文件;2015 年确定拟降为Ⅱ类的共11 项,到2017 年1 月为止,尚未有相关重新分类文件发布。

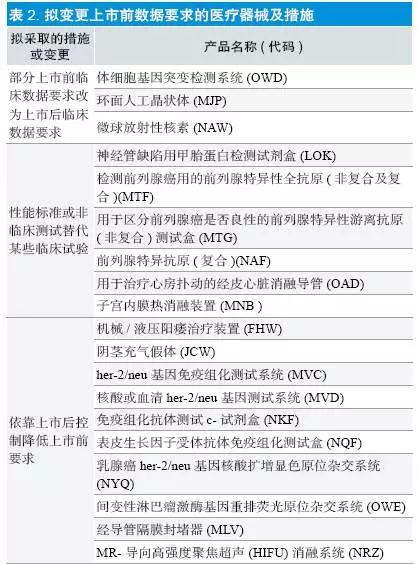
(2)保持管理类别,拟更改数据要求:共27 项,其中FAF/MLV/NRZ/MEQ/MMY/MNB 共六项为2015 年完成部分,如表所示,主要拟采取的措施有:①部分上市前临床数据要求改为上市后临床数据要求;②性能标准或非临床测试替代某些临床试验;③依靠上市后控制降低上市前要求;④参考实际情况,减少上市后研究;⑤其他方式降低或减少上市前临床数据要求,比如将需要大量患者的单组研究改为有预设结束点、需要较少患者的对照研究。其中MJP/MKQ/MNM三个产品的要求变更已于2015 年完成(见表2)。
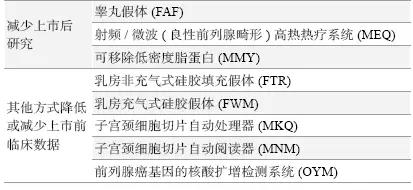
(3)回顾性审核期间,已减少或更改数据要求和/ 或重新分类:2014 年完成3 项,2015 年完成5 项,共8 项。见表3。

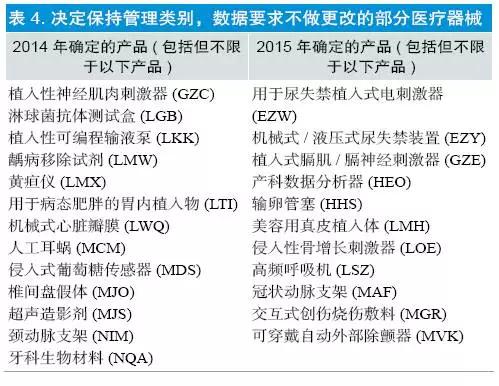
(4)保持Ⅲ类,数据要求不做更改:共涉及产品148 个,2014 年确定96 个,2015 年确定52 个,部分产品见表4。
3.思路解析
美国FDA 的医疗器械监管一向以严格见称,其监管水平获得了全球范围的认可。实际上,确实因其严格的准入措施防止或减少了很多医疗事故在美国发生,FDA 曾撰文“Unsafe and Ineffective Devices Approved in the EU that wereNot Approved in the US[3]”,对该类事件进行了介绍,文中总结了一系列近年来在EU 市场发生严重不良事件的器械,而在美国由于其严格准入,这些器械尚未获批上市,或者已被拒绝进入美国市场。
一直以来,在获得广泛赞誉的同时,美国FDA 这样的监管尺度也面临来自各方面的巨大压力。这种压力主要是来自于行业、消费者对其注册程序繁杂、审批过程过于漫长的抱怨和不满,反对者认为这样的要求使阻碍了医疗器械行业创新和发展,使其成本增加、入市时间延长。相比欧洲地区的患者,美国患者为获得新型医疗器械往往需要等待更长的时间。
为缓解这些压力,FDA 一直致力于在保障安全性的同时,以科学监管手段提高审批效率,降低企业的合规成本,本次对PMA 产品的回顾性审核充分体现了这样的思路和出发点:在对器械临床应用情况进行收集分析的基础上,不断对监管手段的效率、必要性进行探讨,从而做出更为科学合理的调整,使其监管要求更贴近实际,产生更好的社会综合效益。
来源: 本文刊登于《中国医疗器械信息》2017年第11期




